|
 |
|
We are investigating protein-protein interactions for more than 2000 human proteins whose structures are known, with particular focus on those proteins that play a role in neuromuscular diseases. The database of information that will be produced will help researchers design molecules to inhibit or enhance binding of particular macromolecules, hopefully leading to better treatments for muscular dystrophy and other neuromuscular diseases. Phase 1 of Help Cure Muscular Dystrophy has ended in June 2007 and Phase 2 has been launched in may 2009 and ended in the fall 2013. The first and second phase of the project is supported by World Community Grid and by Decrypthon (a partnership between AFM (French Muscular Dystrophy Association), CNRS (French National Center for Scientific Research) and IBM). We are now continuing our investigations on protein-protein intercations within the framework of the MAPPING project (Investissement d'Avenir en Bioinformatique, funded by the French Ministry of Research).
|
| UPDATES for the project |
October 28, 2014 Dear All, Here is some information on how our study of Protein-Protein Interactions (PPI) is evolving within the framework of the MAPPING project (Investissement d'Avenir en Bioinformatique, funded by the French Ministry of Research). We are moving in several directions, all aimed at refining information on protein partnership within the cell that has previously been obtained [Lopes et al 2013; Sacquin-Mora et al. 2008]. The different directions taken by the project are explained below: DOCKING COMBINED TO EXPERIMENTAL INTERFACES
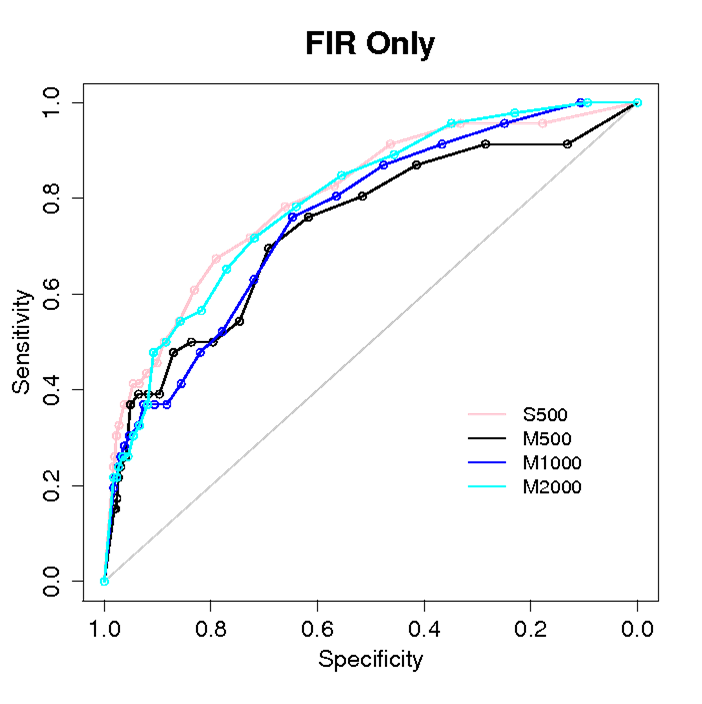
Fig. Prediction of partners among 46 proteins, by combining rigid docking based on geometrical complementarity and knowledge of experimental interfaces. 500 (pink, black), 1000 (blue) and 2000 (cyan) docking conformations have been analyzed. The docking has been realized starting from original PDB positions (S) or from random positions (M). WHEN DOCKING IS CROSSED WITH PREDICTIONS
COEVOLUTION ANALYSIS AND PROTEIN-PROTEIN INTERACTIONS
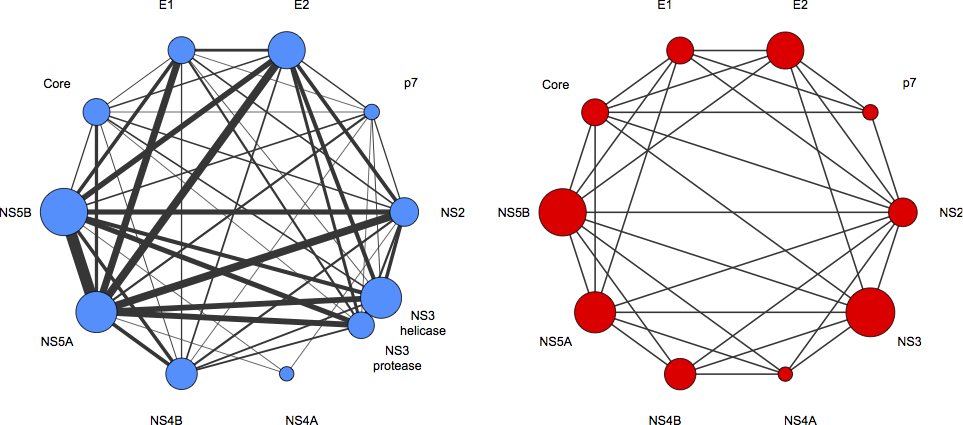
Fig. HCV protein-protein interaction network [Champeimont et al. unpublished results]. The blue network reports the result of coevolution analysis performed on the full HCV polyprotein (for three genotypes). The width of the lines is proportional to the number of predicted direct and indirect interactions (at the domain level), while the circles representing the proteins have an area proportional to the protein length. The red network reports all experimentally known HCV protein-protein interactions. Coevolution analysis is realized with a new version of the BIS tool [Dib&Carbone, 2012].
PROTEIN-PROTEIN INTERACTION AND DOCKING In parallel, R.Lavery's team has developed a new coarse-grain docking approach, using a simplified representation of protein structures, based on the PaLaCe model [Pasi et al. 2013]. This model provides very encouraging results for the prediction of binding affinity constants for the formation of binary protein complexes [N Ceres, unpublished results]. The development of more refined energy terms representing both electrostatics and interactions with the solvent [Ceres et al. 2012] are underway and should lead to further improved affinity predictions. In terms of docking, the team has developed a multiple minimization approach that can deal with the flexibility of the interacting proteins (either limiting movements to side chains, to protein loops at the interface, or treating the complete protein as flexible). The use of internal coordinates (notably torsion angles) makes minimization much more efficient than it would be using Cartesian coordinates.
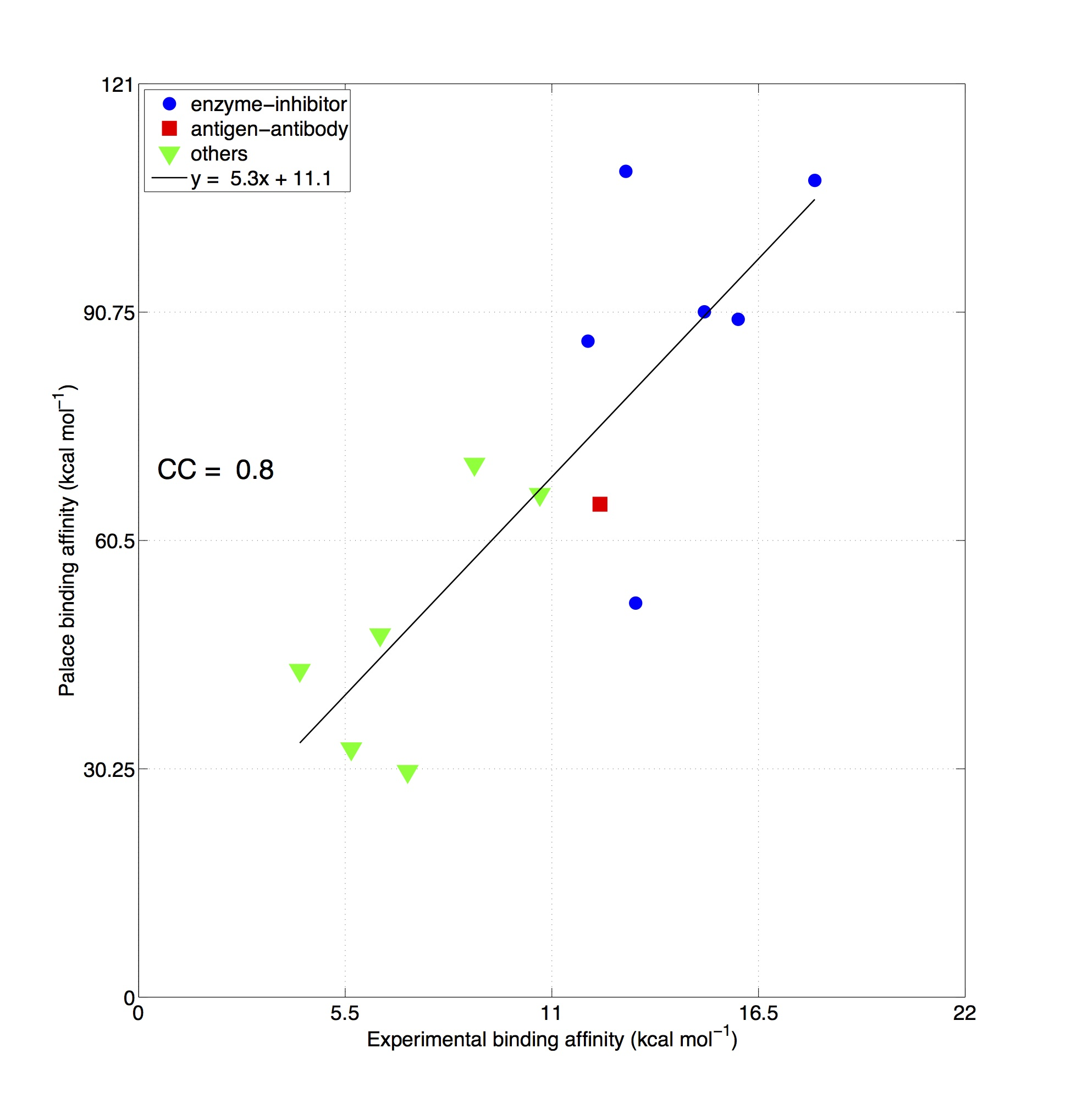
Fig. Comparison between experimentally measured binding affinities of different types of protein-protein complexes (x-axis, data from the "Affinity Benchmark" [Kastritis et al. Protein Science 2013]) and values predicted by PaLaCe (y-axis). The global correlation coefficient is 0.8.
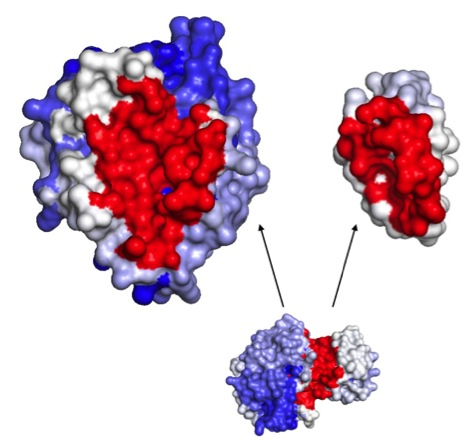
Fig. Docking study between the Alpha Chymotrypsin (left) and the Eglin C (right) proteins, minimized (on 50000 randomly chosen starting points) with the PaLaCe potential. The interaction surface is colored depending on the probability that a residue will participate to the protein-protein interface (red: strong, white: medium, blue: weak). The favored surfaces are in excellent correlations with the experimental structure (visualized on the bottom).
RECENT PUBLICATIONS A.Lopes, S.Sacquin-Mora, V.Dimitrova, E.Laine, Y.Ponty, A.Carbone, Protein-protein interactions in a crowded environment: an analysis via cross-docking simulations and evolutionary information, PLoS Computational Biology, 2013. E. Laine, A. Carbone, Identification of Protein Interaction Partners from Shape Complementarity Molecular Cross-Docking. In A. Petrosino, L. Maddalena, P. Pala (Eds.), IEEE International Conference on Image Analysis and Processing (ICIAP) 2013 Workshops, LNCS 8158, pp. 318–325. Springer, Heidelberg, 2013. A.Carbone, Extracting co-evolving characters from a tree of species. In Discrete and Topological Models in Molecular Biology, N.Jonoska, M.Saito, G.Rozenberg (eds.), Springer, 2013. Pasi, M., Lavery, R., & Ceres, N. PaLaCe: A Coarse-Grain Protein Model for Studying Mechanical Properties. Journal of Chemical Theory and Computation, 9, 785–793, 2013. N. Ceres, M. Pasi, R. Lavery. A Protein Solvation Model Based on Residue Burial. Journal of Chemical Theory and Computation 2012 8:2141-2144.
WEBLECTURE A webLecture was organized by World Community Grid in February 2014. The video in on YouTube.
November 13, 2012 Dear All, We are at the end of HCMD2 and I would like to thank you for the patience and persistence in running our docking program in your machines. The huge amount of cross-docking data that we collected, thanks to you (!), has been for the first time realized. It is a mine of information for our research in protein-protein interactions and it will constitute a precious amount of information also for our colleagues in the world interested in molecular docking. We finished to analyze the data on the 168 protein complexes run on HCMD1 and we now know what has to be done next. We shall integrate novel and quantitative, experimental data on protein binding to predict not only the conformation of interacting proteins, but also which proteins will interact and how strongly. This involves four specific challenges: 1) Obtain quantitative experimental data on protein interactions with a wide range of binding affinities. We will use surface plasmon resonance 2) Use evolutionary sequence data to detect protein residues involved in interaction interfaces and pairs of interacting proteins. We will identify key residues within interaction sites and co-evolution signals between pairs of interaction sites in order to predict interacting partners and integrate this information into a refined molecular docking approach, with the aim of identifying binary interactions within a large set of proteins. This goal will include constructing an automated pipeline for co-evolution analysis of single proteins and protein pairs. 3) Formulate new protein-protein interaction potentials using experimental data, molecular simulations and existing structural data. Molecular simulations coupled with free energy calculations will be used to obtain an atomic-scale view of the dissociation of a limited number of the weak and strong protein interactions studied by microcalorimetry. We will determine the extent to which complexes have well-defined conformations and fully desolvated interfaces. This data will be used to formulate and iteratively refine new interaction potentials within a coarse-grain model, which will be sensitive to binding affinity. 4) Carry out a refined analysis of the large database of protein interactions that you generated (!) to characterize interaction networks and binding promiscuity. During stage two of the Help Cure Muscular Dystrophy project (HCMD2), the resources of the World Community Grid (WCG) were used to dock all possible protein pairs within a set of 2200 proteins, potentially important for understanding and treating The methods and interaction data derived from our studies will be freely available to the scientific community by the implementation of web servers and web databases. We will do all this with a 4 years funding from the French ministry of research that was awarded to our group this year. We shall devote this grant to the development of the new tools (in biophysics and bioinformatics) mentioned above, as well as on the analysis of the HCMD2 dataset to arrive to the best prediction possible on the human protein-protein interaction network that you generated in these two last years. To keep you informed on the development of the project, I shall provide news on the advancements in my webpage. Pointers to the publications will be given there. If by any chance I do not post news from more than 6 months, send me a reminder! THANK YOU again to all of you from all the scientists of the HCMD1 and HCMD2 projects. Best regards to all, Alessandra
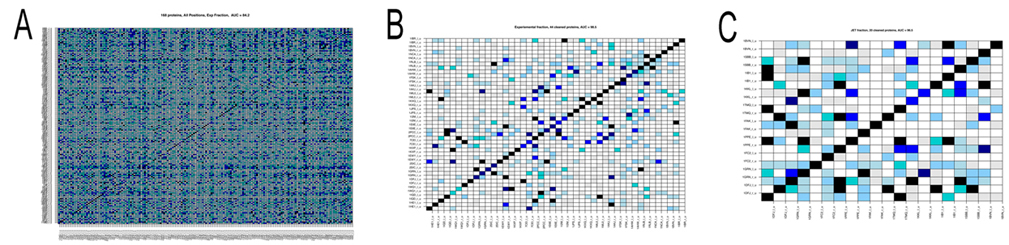
December 21, 2011 Hi to all, with Sophie and Richard we have written up an account of the docking analysis of the 168 protein complexes of the Mintseris dataset tested in phase 1. The paper is under review right now and will give you the link as soon as it is published. The analysis of the dataset of 168 protein complexes is not finished yet! In fact, we try to improve the signals for the detection of partnership. There are two main points that one needs to keep in mind. In phase 2 we do not know the real partners and we had to use predictions of interaction sites to run MaXDO. This was because the search space on a protein surface could not be exhaustively explored, even with the help of WCG. it would be far too big! This means that we need to understant on a pool of proteins that we know (that is the 168 protein complexes) how the predictions of protein interaction sites will impact partners predictions. This is what we carefully investigate right now. It takes time! There are a number of intermediate results that you might like to know about: 1. the analysis realized in [Sacquin-Mora et al. 2008] on 12 complexes, has been scaled to 168 complexes, and it highlighted a predictive protein-protein interaction power of AUC=0.84 (see Figure A below) when using knowledge on real interaction surfaces and when exploring the whole protein surface. It is important to stress that this successful scaling of the analysis in [Sacquin-Mora et al. 2008] to 168 proteins was not an obvious guess! Why successful? The AUC is a probability measure used to evaluate the accuracy of the test. Values vary from 0 to 1, where 1 represents a perfect test and 0.5 represents a worthless test. Roughly speaking, one can think of the following ranking: 2. We also observed that amongst the 168 protein complexes several had the tendency to bind to nearly all other proteins and others showed very few strong interactions. Both these families of proteins negatively contribute to partnership prediction, and, when eliminated, enable the predictive power to be increased to an AUC=0.98 (see Figure B). 4. Nevertheless improvements are still required, since when JET predictions are used to delimit the docking area, as well as to compute the numerical index that discriminates partners, the predictive power falls at an AUC=0.59. This implies that better interaction patch detection has to be developed. However, we note that a subgroup of 20 complexes was identified where JET predictions already yielded very good predictions (AUC=0.97; Figure C below), suggesting that generating subgroups by categorizing protein interaction proclivities could improve performance.
Figure. Matrices of pairwise interaction indexes for different subsets of proteins. High interaction scores (between 0.7 and 1, blue and black in the color scale) indicate a high probability of interaction. Proteins are ordered in the matrix such that true interacting partners lie on the diagonal. A: full dataset of 168 protein complexes. Interaction scores were computed using knowledge of the experimental interfaces (AUC=0.84). B: subset of 44 protein complexes leading to an AUC=0.98. Interaction scores were computed using knowledge on experimental interfaces. C: subset of 20 proteine complexes leading to an AUC=0.97. Interaction scores were computed using interfaces predicted by JET. At the moment we work on coevolution between protein interfaces and on improving JET interaction predictions. With both advancements we count improving identification of new partners, and increase the AUC above. We have done a lot of work already on this. A new approach to coevolution analysis, treating especially conserved sites like protein interfaces, has been recently developed at the lab. More on this soon. Merry Christmas and a Happy New Year to all! Alessandra
July 31, 2010 Hi to all! Thought to try to explain what we are doing right now before you take some vacation, like the scientists here. Hope it will help to feel that things are improving and that the project is very active from this side!! Actually, someone new will join the group on september, Anne Lopes. Anne is assistant professor in structural bioinformatics and has a background in physical-chemistry. She is very interested in working on the protein partnership problem with the numerical approach we developed and on the data analysis of the huge amount of information you are producing! The state of the art here is the following. In the paper [S. Sacquin-Mora, A. Carbone and R. Lavery (2008), Identification of protein interaction partners and protein-protein interaction sites, J. Mol. Biol. 382, p1276-1289] we developed a numerical method to detect protein partners. The method was presented and tested on a small quantity of known protein complexes. As you can imagine, as soon as the data from HCMD Phase 1 arrived (THANKS TO YOUR CONTRIBUTION!!) we retested the approach to verify whether we could confirm the results on a larger dataset. This is indeed the case, the method works, and we can distinguish protein partners within the about 150 proteins tested. We observed that the signal is much less sharp when we work with 150 proteins than with 12 proteins (like in the paper) though and that some extra work should be done to improve the numerical method. Remember that for HCMD Phase 2 we shall search for partners among about 2200 proteins. At the moment, we have improved the formula introduced in the paper and we are developing an "intelligent" approach to arrive fast and surely to identify a small number of potential partners for any protein. Let me give you an insight on the complexity underlying the problem. It has something to do with the understanding of protein population. This is an important point to assimilate, if you like to understand a bit more of our analysis. When we consider a protein, we do not just study one protein (that is, its geometry and its physico-chemical properties: this is already taken into account in the docking algorithm running on your computers and into JET, the program that allowed us to predict protein binding sites) but we rather study its behaviour with the population of proteins that are around it (in the cell; for the HCMD phase 2, population means the 2200 proteins analyzed in your computers). In other words, when we look at a protein we hope to get a signal on its partnership by looking at her way to interact with all other proteins in the population. This means that we hope to learn from bad interactions as well as from good interactions. The information that YOU are giving us provides to us some insight on what is bad and what is good! but this is not enough and we shall use also some extra observation on the interaction of the protein within a population. Some proteins are slippery, meaning that they do not seem to glue to any partner. Some others are gluing, meaning that they do glue to essentially everybody. Then there are many other proteins (about a half) that seem to stick on the right place with some specificity. They are the easiest to study. When we use, in our calculations, contributions coming from the entire population, one should think that these contributions come, in principle, from slippery proteins, gluing proteins and many other proteins whose behaviour is less sharply characterizable. "Noise" might enter into the calculation and we wish to reduce it. Learning from the whole set of interactions of a protein, means to learn to which group the protein belongs to. Once this is determined, the numerical criteria that we developed could be adjusted to accurately predict a partner or a small set of potential partners, whenever possible. The understanding of the whole set of behaviours that we need to take into account to know how to correctly evaluate the data coming from WCG is our goal today. There are a few other concerns that are present in our analysis, and they have something to do with : 1. the algorithmic aspects concerning the handling of large amount of information to be combined for the "learning" approach I mentioned above. 2. the fact that on HCMD phase 2 data analysis, we use JET predictions of protein interaction in our numerical criteria instead of actual real interfaces as done in the paper cited above. This implies a loss of precision that we should consider in our numerical evaluations of the interactions. These informations should give you some insight on the complexity of the question we face today. Hope that everyone will be feeling that we are advancing, together, for a project that runs alive and hopefully will reserve exciting surprises to all. We expect it. Have a good summer! Alessandra |
| Help Cure Muscular Distrophy Project and the World Community Grid |
Computational grids are emerging as a new paradigm for sharing and aggregation of geographically distributed resources with the aim of solving large-scale computational and data intensive problems in science. This project proposes to apply this powerful computational schema to the detection of protein-protein interactions. Identifying pairs or larger complexes of functionally interacting proteins, or determining the binding of a protein to a DNA sequence or to a ligand are fundamental problems in biology with immediate consequences in drug design. This multidisciplinary project directly addresses this question by setting the goal of screening a database containing thousands of proteins, predict functional sites involved in binding to other proteins or ligand targets, and determine whether two proteins are potential interacting partners in the cell. The project will determine information on the structure of macromolecular complexes which is important not only for identifying functionally important partners, but also for determining how such interactions will be perturbed by natural or engineered site mutations in either of the interacting partners, or as the result of exogenous molecules, and, notably, pharmacophores. A database of such information would be of significant medical interest since, while it now becomes feasible to design a small molecule to inhibit or enhance the binding of a given macromolecule to a given partner, it is much more difficult to know how the same small molecule could directly or indirectly influence other existing interactions.
Given n protein structures, they are docked one against the other, that is cross-docked. Molecular modeling refers to theoretical methods and computational techniques to model or mimic the behavior of molecules. These methods and techniques are used to investigate the structure of biological systems such as protein folding or molecular recognition of protein-ligand binding, ranging from small chemical systems to large biological molecules and assemblies of material (protein complexes). Protein-ligand docking is a molecular modeling technique to predict the position and orientation (the 3D-structure) of a protein in relation to a ligand (another protein, DNA, drug, etc.). Docking methods are based on purely physical principles; even proteins of unknown function (or which have been studied relatively little) may be docked. The only prerequisite is that their 3D-structure has been either determined experimentally, or can be estimated by some theoretical technique. The docking approach generally starts with a database of known molecules and attempts to find pairs of molecules which have an affinity to bind to one another. The affinity is estimated using a so-called scoring function. In the end, a list of the best-binding molecules for a targeted protein is returned. The quality of fit has a geometric and a chemical component. The geometric component measures how well the surface shapes (the 3D-structures) complement each other like a hand in glove. The chemical component measures the quality of the atomic interactions between the partner molecules (i.e. are the interactions strong or weak?). For complex structures like proteins (the smallest are composed of hundreds of atoms), it takes considerably computer time to determine the fit of correct protein-protein interactions. Without World Community Grid, the computations required to conduct the docking was prohibitively time consuming. For the first 168 selected proteins, the estimated CPU time on a 2 GHz PC should have been about 8,000 years. A solution to this computational barrier is to use evolutionary information to predict potential binding sites and realize localized docking only on surfaces which are most likely to interact. By using the prediction of protein binding sites, based on protein evolution, highly reduces computational time by a factor of 100 and therefore allow us to extend the analysis at large scale with the crucial help of World Community Grid. Without World Community Grid, the computations required to conduct the (localized) docking at large scale would be also prohibitively time consuming. Volunteers donating their computer time to World Community Grid searched (in phase I) and will search (in phase II) for the best protein-protein partners. The main pool of calculations
runs on World Community Grid: Preliminary calculations
and a posteriori analysis run on Decrypthon Grid and on Grid'5000: |
Participants : teams and computing infrastructure |
Grid computing: Jean-Marie Chesneaux team, Laboratory of Computer Science (LIP6), UMR 7606 CNRS-UPMC, Université Pierre et Marie Curie, Paris. Genetic analysis of myopathies: Pascale Guicheney team, INSERM Laboratory U582, Myology Institute, "Pitié-Salpetrière" Hospital, Paris. Molecular modelling: Richard
Lavery team, Intitut de Biologie et Chimie des Protéines, UMR 5086 CNRS-Université de Lyon, Lyon. Thanks to the funding of AFM and CNRS, three postdocs actively partecipated from 2005 to 2008 to the advancement of the project : Stefan Engelen - IR Genoscope, Evry
Yann Ponty - Postdoc at Laboratoire
d'Informatique de Paris 6, CNRS-UPMC Sophie Sacquin-Mora - CR CNRS - Laboratoire
de Biochimie Théorique, UMR 9080 CNRS, Institut de Biologie Physico-Chimique,
Paris Sophie Sacquin-Mora and Yann Ponty are working actively on the project even if associated to other institutions now. Decrypthon Program provided the infrastructure necessary to assure the portability of the software to the Decrypthon Grid and to WCG. The Laboratoire d'Informatique du Parallelisme (ENS, Lyon), partner of AFM/CNRS, gave us the possibility to run tests and a posteriori analysis on the French University Grid5000. For this, we acknowledge the work of Raphael Bolze (LIP, ENS Lyon). Nicolas Bard (LIP, ENS Lyon) and Michael Heymann (LIP, ENS Lyon) will soon replace Raphael on the same tasks. The project run on WCG. Thousands of internautes offered their computer time to the project. To them we are most grateful.
|
| What have we done on Phase 1 |
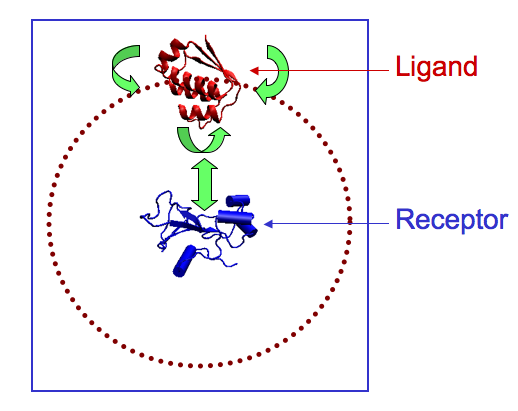
In Phase
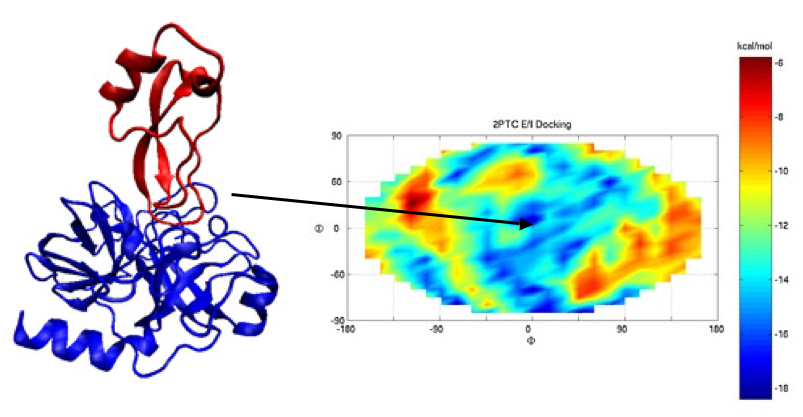
1 we tested the feasibility of the docking algorithm MAXDo on a database of 168 proteins for which we had crystallographic information and experimental evidence of the interaction of these proteins complexes in the cell. We performed cross docking on all protein pairs (see figure below on the left where the receptor is fixed at the center of the sphere and the ligand moves all around the ideal sphere indicated with red dots) and collected energy maps (with the Euler angles θ and ? along the vertical and horizontal axis; see an example of the maps issued from our calculations on the right below) after docking. For each map, the experimental binding site of the receptor in its complexed form with the ligand is located at the center (see complex in the middle of the figure below). Blue and red areas correspond to the most negative and the least negative energies, respectively. These data were then analyzed and crossed with a numerical criteria that we had developed for discrimination of protein partners. In the figures below you can see the docking schema, a complex and the energy map associated to the complex, computed on WCG.
|
| HCMD status report, february 2009 |
From the end of Phase 1 our efforts have been concentrated on four directions: the finalisation and testing of JET, the analysis of data gathered in phase 1, the interface between JET and MAXDo, the algorithmic improvement of MAXDo, the constitution of a database of proteins that will be analysed in phase 2. We completed the essential parts of all steps and we are ready to start phase 2 of the project.
|
| 1. Finalisation of JET and test of its performance |
Information obtained on the structure of macromolecular complexes is important for identifying functionally important partners, but also for determining how such interactions will be perturbed by natural or engineered site mutations. Hence, to fully understand or to control biological processes we need to predict in the most accurate manner protein interfaces for a protein structure, possibly without knowing its partners. Joint Evolutionary Trees (JET) is a method designed to detect very different types of interactions of a protein with another protein, ligands, DNA and RNA. JET uses evolutionary information, namely how certain positions within the primary sequence of a protein are conserved in a given protein family, and also physico-chemical properties of residues to finally predict the location of protein-protein interface sites on the protein surface. It uses a carefully designed sampling method making sequence analysis more sensitive to the functional and structural importance of individual residues, and a clustering method parameterized on the target structure for the detection of patches on protein surfaces and their extension into predicted interaction sites. JET is a large scale method, highly accurate and applicable to search protein partners. JET has been conceived and developed in different stages along 4 years of work which now finalized in a publically available software package and a publication [Engelen et al 2008]. The information obtained with JET is extremely interesting for the HCMD project because we can save a large amount of computational time by restricting the search of the conformational space between the two proteins to the area surrounding the predicted interface. The data resulting from phase 1 of our project were used to adjust various parameters of the JET program and see how far we could go in filtering the starting positions in the docking calculations without loosing relevant information on protein interactions.
|
| - Read more on JET |
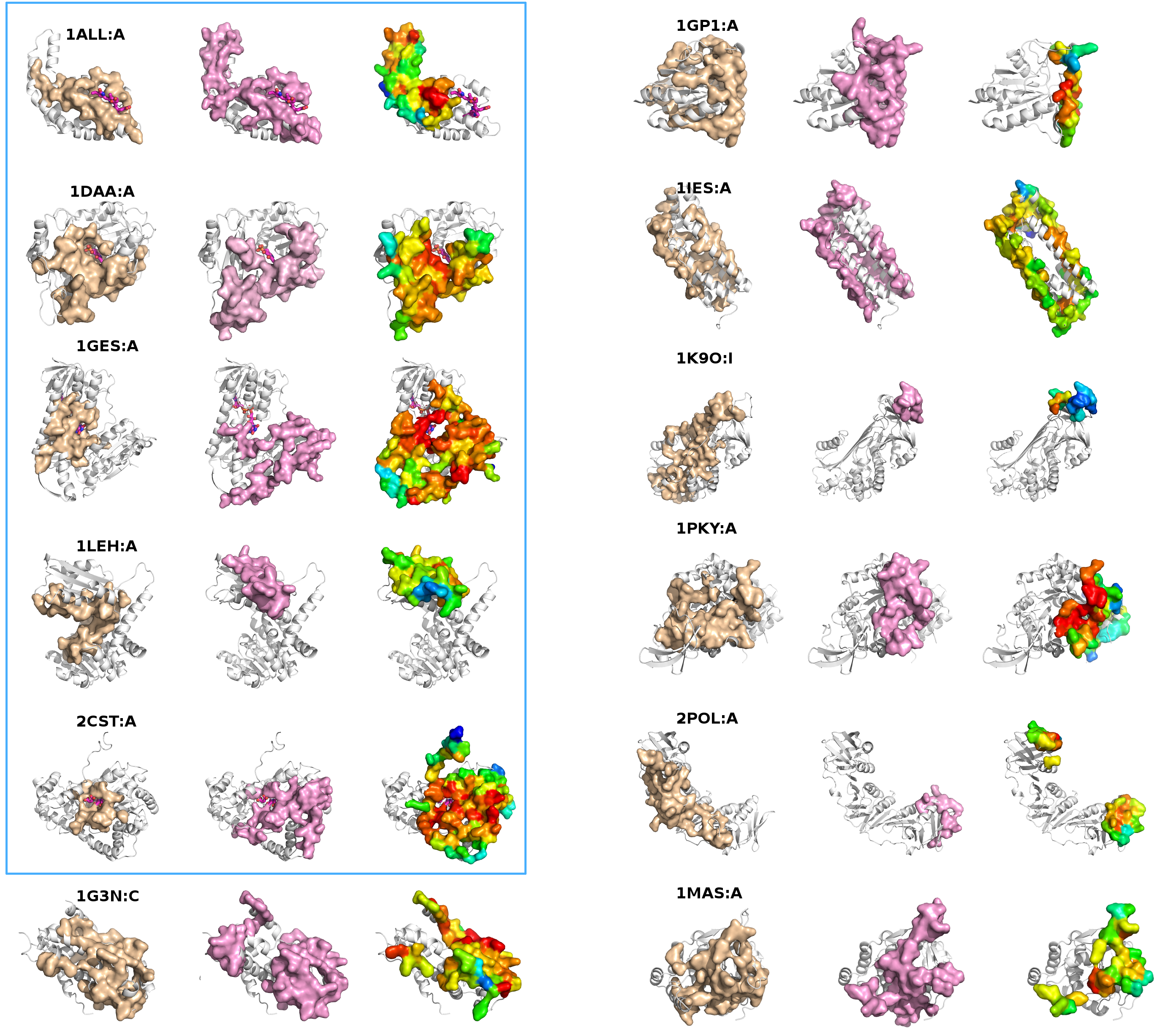
The Joint Evolutionary Trees (JET) method detects protein interfaces, the core of residues involved in the folding process, residues susceptible to be relevant to site-directed mutagenesis and to molecular recognition. The approach, based on the Evolutionary Trace (ET) method, introduces a novel way to treat evolutionary information. Families of homologous sequences are analyzed through a Gibbs-like sampling of distance trees to reduce effects of erroneous multiple alignment and impacts of weakly homologous sequences on distance tree construction. The sampling method makes sequence analysis more sensitive to functional and structural importance of individual residues by avoiding effects of the overrepresentation of highly homologous sequences and it improves computational efficiency. A carefully designed clustering method is parameterized on the target structure to detect and extend patches on protein surfaces into predicted interaction sites. Clustering takes into account residue's physical-chemical properties as well as conservation. Large-scale application of JET requires the system to be adjustable for different datasets and to guarantee predictions even if the signal is low. Flexibility was achieved by a careful treatment of the number of retrieved sequences, the amino acid distance between sequences, and the selective thresholds for cluster identification. An iterative version of JET (iJET) that guarantees finding the most likely interface residues is proposed as the appropriate tool for large scale predictions. Tests are carried out on the Huang database of 62 heterodimer, homodimer and transient complexes, and on 265 interfaces belonging to signal transduction proteins, enzymes, inhibitors, antibodies, antigens and others. A specific set of proteins chosen for their special functional and structural properties illustrate JET behavior on a large variety of interactions covering proteins, ligands, DNA and RNA. JET is compared at large scale to ET, and to Consurf, Rate4Site, siteFiNDER|3D, SCORECONS on specific structures. A significant improvement in performance and computational efficiency is shown.
Fig 0. JET prediction on the interaction between a RNA strand and a protein complex: the crystal structure of a complex of TRP RNA-binding attenuation protein with a 53-base single stranded RNA containing eleven GAG triplets separated by AU dinucleotides (Antson, A.A., Dodson, E.J., Dodson, G., Greaves, R.B., Chen, X., Gollnick, P. (1999) Structure of the trp RNA-binding attenuation protein, TRAP, bound to RNA. Nature 401: 235-242 ). Residues are colored from red (most conserved) to blue (no conservation), and red-like residues indicate predicted interacting residues. Notice the red color of an entire face of the complex (left) and the blue color of the opposite face (middle). The RNA is in contact with the red surface (see right view).
|
| 2. Analysis of data coming from Phase 1 |
Phase 1 generated a huge amount
of data on protein-protein interactions, which we are still working
on. This data gives us valuable information on the way proteins
interact. |
| - Identification of protein interaction partners |
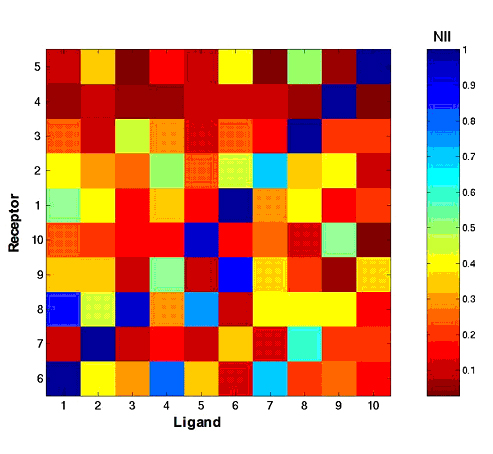
In order to evaluate the quality of the protein interactions modelled with the MAXDo program (which ran on WCG during phase 1 of the HCMD project) we developed an Interaction Index (II), based on the interaction energies between proteins and the residues found at the protein interface. The II value ranges from O (no complex formation) and 1 (excellent interaction between the two proteins). If we compare the distribution of the II value for all protein pairs (over 28,000) and only pairs of experimental partners (168) we see that "real partners" usually form complexes with a significantly larger II than randomly chosen partners: The average II value is only 0.18 for the whole database, while the "real" couples yield an average II of 0.32. There are also some very important variations depending on the type of complex considered. For example, in a reduced dataset excluding enzyme-inhibitor complexes, the experimental partners had a remarkable II average value of 0.91, which make then very easily distinguishable from the random partners (see Fig.1).
Fig. 1: Normalized Interaction Index matrix for a reduced dataset of ten proteins. The rows and columns are ordered so that experimentally observed complexes lie on the trailing diagonal of the matrix. These complexes clearly distinguish themselves, with a higher II, from the incorrect off-diagonal complexes.
|
| 3. Interface between JET and MAXDo |
Since JET is used to restrict docking interaction targets, it was critical to assess the sensitivity of our tool in order to ensure a reasonable coverage and not overlook entire interactions. To that purpose, we tested the software on two large sets of proteins originally used for benchmarking docking in silico approaches (Mintseris 2.0 + Kanamori). In some case, high sensitivity/specificity couples can be observed and JET pinpoints the interaction site (See Fig. 2). More generally, we observed an average 38% sensitivity with a remarkable typical specificity of 80%, under the settings used for restricting MAXDo's conformational space. This means that a bad prediction is in most cases, an absence of prediction, which can be detected and corrected prior to passing the protein to our docking software MAXDo. Furthermore, large discrepancies are observed for the sensitivities achieved on proteins having different functions. For instance, JET performs very well on Enzymes substrates/inhibitors (55.3% Sens. 75% Spec.) and rather poorly on Antigens/Antibodies (17% Sens. 81% Spec.). We will use this information to adapt our restriction of MAXDo's conformation space, being more stringent for easy family and more relaxed for hard ones. Despite these corrections, we expect the MAXDo/JET joint approach for an ab initio cross-docking to perform better on Enzymes than on Antigens/Antibodies.
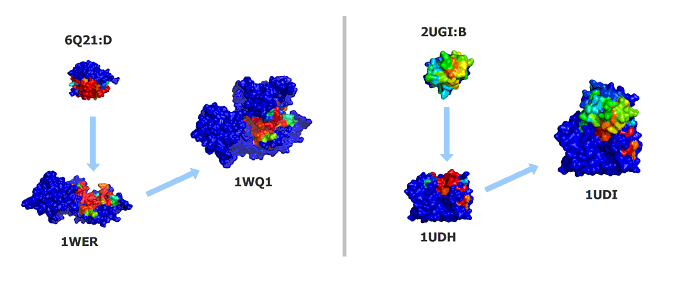
Fig. 2: For both the Ras-RasGAP complex (PDB:1WQ1, left) and the DNA glycosylase bound to its inhibitor (PDB:1UDI, right), JET accurately targets the experimentally-observed interfaces. More precisely, the (sensitivity,specificity) couples achieved by JET on individual chains are: (74%,89%) for 6Q21:D, (58%,95%) for 1WER, (52%,77%) for 2UGI:B and (65%,89%) for 1UDH. Such figures are not only suitable for restricting MAXDo search space, but will also greatly help directing our search for the real docking sites during a posteriori analyses.
|
| - A posteriori analysis |
A preliminary analysis of the data produced during phase 1 showed that Interaction Indices, recalculated using JET-predicted data instead of experimental ones, still carry some discriminative power. We have good hope that taking into account more features on the candidate interfaces, combined with automated machine learning approaches, will greatly enhance the quality of our analysis of the second phase results. More specifically, supervised learning procedures such as Support Vector Machines (SVM) will be fine-tuned during the second phase results to automatically distinguish between positive (Interacting proteins) and negative (Non-interacting proteins) examples.
|
| 4. A new version of MAXDo |
We also worked on a faster, more efficient version of MAXDo. This new version takes into account the information produced by the JET program concerning the location of protein interfaces, so that we now need to test only around 15% of the starting positions for docking that we previously studied with MAXDo (see Fig. 3). Combined with other improvements made to MAXDo, the docking of a protein pair should take only 3% of the computation time that was necessary during Phase1.
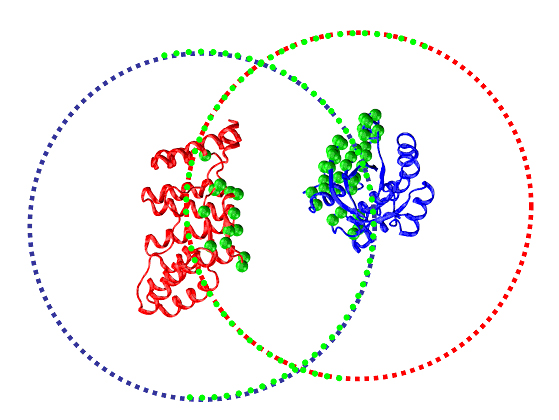
Fig. 3: Cartoon representation of the RacGTApase/P67Phox complex (pdb code 1E96). The systematically generated starting positions for the receptor and ligand proteins are plotted as blue and red points. After filtration using JET information, docking calculations are only performed for those starting points that are located in the vicinity of JET predicted interface residues (plotted as green spheres on the proteins), thus considerably reducing the computation time.
|
| 5. Proteins list that will be analysed on phase 2 of the HCMD project |
In phase 2, we will profit from this speedup to work on a larger, but also more targeted protein database. It will include around 2200 human proteins, 200 of which are of interest for neuromuscular diseases and have been proposed as targets for study by the medical research groups working with us in this project. About 200 more are structural models with potential interest for muscular dystrophy. The massive docking experiment performed during phase 2 will give us precious information on how these target proteins interact within the human body (about 1800 protein structures with yet unknown involvement in muscular dystrophy will be analysed), helping biomedical researchers designing new strategies to neutralize them and develop therapies for neuromuscular diseases.
|
| - To know more on the list of proteins |
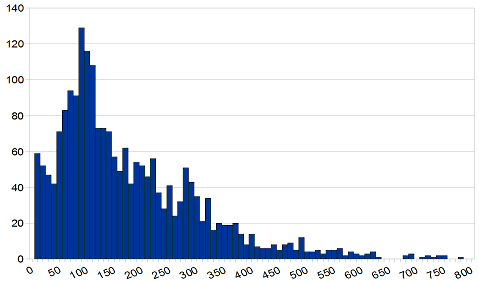
The list of proteins considered for phase 2 was initially selected by experts on three major classes: Neuromuscular diseases, experimentally-determined (Guicheney and Carbone labs) and predicted structures (Laboratoire de Bioinformatique et Genomique Integratives, IGBMC); others (like heart and brain-related) experimental structures (Carbone lab). This initial list was later semi-automatically filtered to weed out very similar proteins. To that purpose, we used the ASTRAL structurally non-redundant database, and added a final filter based on sequence similarity (PDBSelect95 database) in order to cover as much of the structural and functional diversity as possible. Overall, 2263 single-chain proteins of lengths ranging from 25 to 800 residues will be run on the grid (See Fig. 4). Since our ability to run MAXDo on such a large dataset relies on an initial restriction of the conformation spaces, we have installed JET on the Decrypthon grid in Lyon. At the moment, JET is being run simultaneously on the machines of the grid and has already yielded results for more than half of the proteins. Full results should be available during the upcoming weeks. These results will be carefully post-processed, using conclusions from our analysis of the first phase, before we feed the JET-annotated PDB files to MAXDo.
Fig. 4: Distribution of the number of residues (x-axis) for the proteins (y-axis) in the list that will be investigated during phase 2 of the HCMD project.
|
| Publications |
S. Sacquin-Mora, A. Carbone and R. Lavery (2008), Identification of protein interaction partners and protein-protein interaction sites, J. Mol. Biol. 382, p1276-1289. S. Engelen, L.A. Trojan, S. Sacquin-Mora,
R. Lavery and A. Carbone (2009), Joint Evolutionary Trees: a large
scale method to predict protein interfaces based on sequence sampling,
PLoS Comp. Biol. 5(1): e1000267. doi:10.1371/journal.pcbi.1000267. |
| Some numbers |
HCMD2 studies cross docking of 2246 human protein structures. In total we shall dock 2 466 753 pairs of proteins among the 2246^2 possible ones. This means that 913 627 781 945 docking initial positions should be computed by a full cross-docking. By using JET, we can reduce the docking space of 85%, that is we shall have only 137 652 178 995 conformations to be analyzed. Phase 1 of the project studied 28 224 pairs (168 proteins) and explored a total of 10 391 124 240 conformations, that is 13,25 times less than what we shall have to compute in Phase II (in terms of docking initial positions). |